

Large phylogenies comprising hundreds or thousands of individual species are often assembled using sequences from a small number of genetic loci available in online databases such as Genbank. This can be problematic because the phylogeny is limited by the number of loci available and because users must trust the accurate taxonomic identification of sequences that are frequently not linked to specific specimen vouchers such that their determinations could be confirmed.
Herbaria represent a huge and under-used resource for the sequencing of markers from rare and uncommon specimens, and have the advantages of reliable, verifiable determinations and readily available morphological information. However, a few drawbacks of herbarium specimens for this type of work are the highly fragmented DNA that voucher specimens will typically yield, and the labour involved in moving a large number of samples from voucher to lab to complete sequence.
In a new article published in Applications in Plant Sciences, lead authors Ryan A. Folk and Heather R. Kates and colleagues present an integrated management system for streamlining the entire sampling to sequencing pipeline. Called SLIMS (Specimen-to-Laboratory Information Management System), the system uses unique identifiers and a taxonomic database linking the sample to specimen images and wet lab results. Once sampled, linked voucher images are uploaded to the citizen science platform Notes from Nature, where metadata is generated via label transcription, while the tissue itself undergoes a high-throughput DNA extraction and sequencing protocol optimized for herbarium specimens.
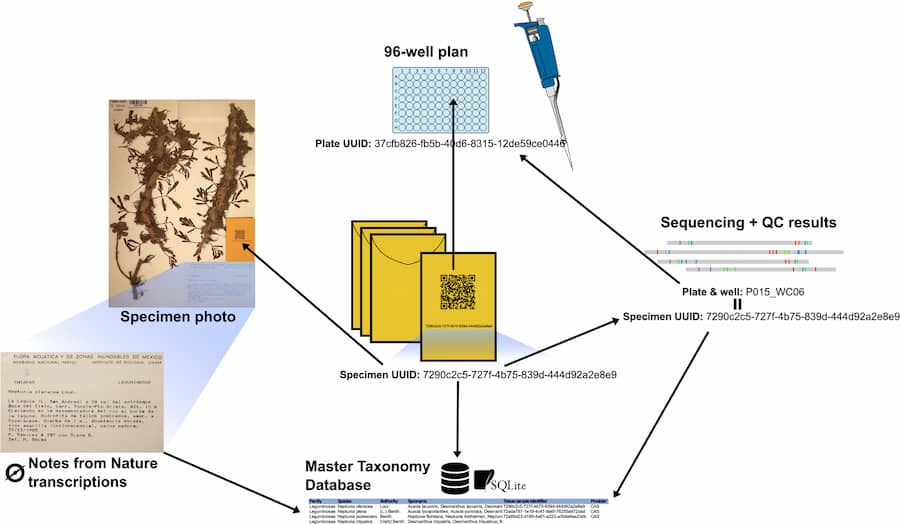
The authors applied their management pipeline to a roughly 15,000-species phylogeny of the nitrogen-fixing clade of angiosperms, producing a dataset comprising around 50% of all species in the clade. Overall, usage of the management system led to herbarium sampling taking roughly 10 person-minutes per specimen and DNA extraction taking around 5 person-minutes per sample. The sampling error rate came to approximately 1.2% and the sequencing failure rate was only 0.2%.
The authors optimized the pipeline for their specific phylogenetic needs, but have offered it as a series of modular scripts rather than a single unified piece of software so that it can be easily adapted to the needs to various projects and sample types. “Considerable work has been dedicated to high-throughput digitization workflows in herbaria; parallel methods to enable other downstream analyses of herbarium specimens may one day enable much of today’s collections to be associated with molecular and other data reliant on destructive sampling,” they write. “We anticipate that high-throughput sampling approaches like that presented here will be a standard part of the phylogenomics toolkit in future large-scale projects.”




