

A gene regulatory network (GRN) describes the hierarchical relationship between complexes of transcription factors, their target genes, and the encoded gene products that then control cellular activity. Understanding GRNs is the key to guiding plant improvement through gene manipulation.
Identification of transcription factors that control signaling and/or developmental responses is challenging because of the large number of putative transcription factors (TFs) that still have no known function, and the fact that TFs which have already been characterized in one context may have additional roles in yet unknown contexts. Additionally, previously characterized TFs likely work in cooperation with other TFs whose function has not been previously described.
A recent paper published in in silico Plants presents TF DEACoN, a new tool that uses previously-published and publically-available Arabidopsis DAP-Seq data to make predictions about which TFs may be involved in an observed transcriptional response. TF DEACoN analyzes data across groups of co-regulated genes to “screen” for transcription factors that may control their concurrent changes in expression.
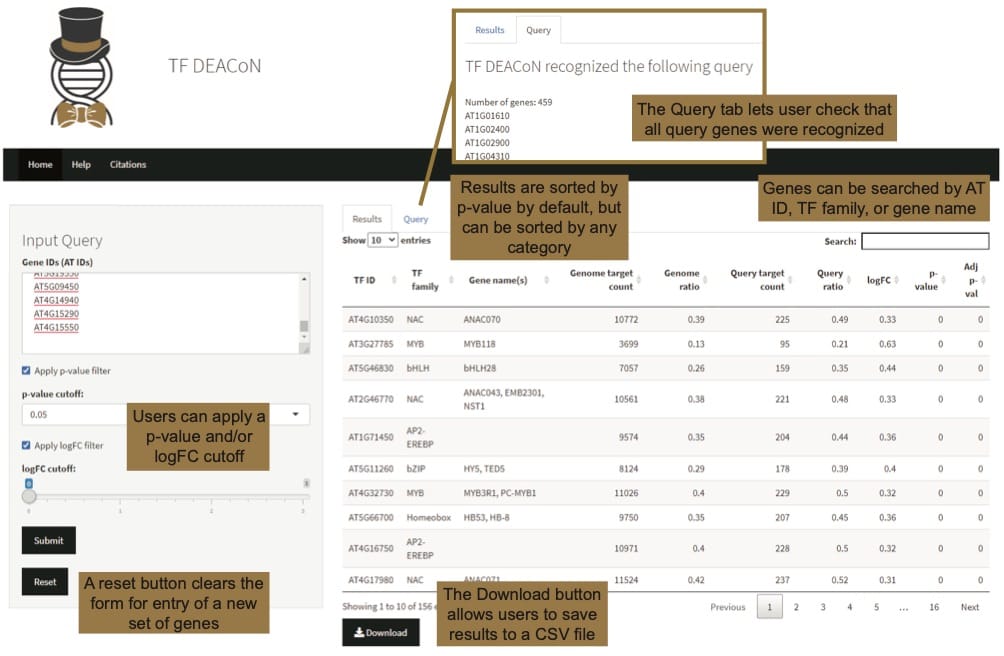
TF DEACoN is publicly available as a user-friendly web app. “The idea behind the analysis is analogous to Gene Ontology (GO) enrichment; instead of functional annotations we’re looking for TFs that target the input genes, and comparing the ratio of targets in this query group to the ratio of targets in the entire genome. We tried to make the app easy to use for researchers. If you have a list of genes that are co-regulated in your process of interest, you simply paste the gene IDs into an input query box, and the tool outputs transcription factors (TF) whose targets are enriched in your set of genes, along with their families and the gene’s AT ID for download,” says Alexandria Harkey, a Postdoctoral Researcher at Wake Forest University.
The authors demonstrated the utility of clustering co-regulated genes based on the similarity of their transcript abundance across time and space to find potential regulators that are evident only when clusters of co-expressed genes are examined. They used TF DEACoN to analyze transcriptional data from roots treated with the ethylene precursor 1-aminocyclopropane-1-carboxylic acid (ACC) to increase ethylene levels, revealing TFs that may act downstream of this hormone signaling to control changes in root development. They then used a genetic approach to show that a mutation in one of the predicted, and previously unknown, transcription factors reduced the negative regulation of lateral root development by ACC.
The combination of filtering and TF DEACoN presented in this paper can be applied to any group of co-regulated genes to predict GRNs that control coordinated transcriptional responses.
The tool “TF Discovery by Enrichment Analysis of Co-expression Networks” (TF DEACoN) was developed using R Shiny and the shinyBS package, and is shared under an open source license at https://github.com/aharkey/TFDEACoN.




